News & Articles

Latest in 2020: Sarcoma (Cancers of the Bone & Soft Tissues) Q&A
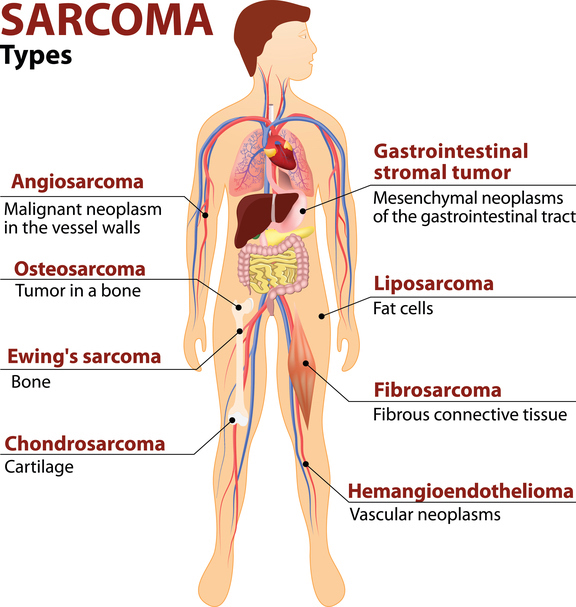
Question 1: What is the survivor rate for Sarcoma and how high is the recurrence rate?
To answer this questions, first let’s recall that sarcoma is not one single disease but an umbrella of more than 70 distinct diseases composed of various sarcoma subtypes.
The natural history of each of these sarcoma subtypes differ; some are aggressive while others are indolent tumors with slow pace of growth and may not spread.
Thus, the prognosis of each sarcoma would depend on many factors, namely, the sub-type of sarcoma, the stage at which it was found, the size and location of the tumor, whether it can be completely removed through surgery and whether adjuvant (preventive) treatment such as radiation or chemotherapy had been administered.
Generally, for the most common aggressive bone sarcoma, take for example osteosarcoma, if the disease was found at an early stage without spread, the tumor could be removed completely and the patient was given an appropriate regimen of chemotherapy, the long term remission rate is approximately 70%.
For aggressive high grade soft tissue sarcoma, if localized (no spread of tumor) and completely removed, the chance of recurrence could be anywhere between 30-50%
Question 2: How promising is immunotherapy for all forms of Sarcoma? more than all people affected by cancer usually use sarcoma therapy as a shortcut. because the cost is cheaper and do not have to always consume drugs
There is only limited clinical research, involving a relatively small number of patients, done using immunotherapy in patients with bone and soft tissue sarcoma. Thus no firm conclusions can be drawn at this point in time. We see pockets of sarcoma subtypes that may possibly respond to immunotherapy, such as undifferentiated pleomorphic sarcoma and liposarcoma. While in bone sarcoma, the initial results observed in trials showed only low responses to immunotherapy. Thus, more research is needed in this field; chemotherapy remains the standard of care.
At this point in time, immunotherapy would be considered an experimental therapy in sarcoma until more data emerges in this field. Sarcoma patients are encouraged to participate in clinical trials if one is available to further answer these clinical questions. Any decision to consider immunotherapy in sarcoma would have to be made after discussions with your healthcare provider.
Question 3: Possible to mistake sarcoma with other cancer during Biopsy or Immunohistochemistry test? will core needle biopsy sufficient to test if doctor suspects either sarcoma or lymphoma? Is Bcell lymphoma a Sarcoma?
In Sarcoma, histo-pathological diagnosis is key. In short, getting the right diagnosis is half the battle won. Before we initiate any form of treatment, I always insist on getting a biopsy or at the minimum review the samples that had been previously taken. A biopsy involves removing a small sample of tumor tissue for analysis. This analysis should then be performed by an EXPERT SARCOMA PATHOLOGIST who is familiar with sarcoma. The reason for that is that sarcoma is a rare and diverse tumor. Studies have consistently shown that up to 20% of sarcoma diagnoses may be discordant when read by non sarcoma pathologists vs expert sarcoma pathologists.
A core needle biopsy is usually sufficient to obtain tissue for analysis. Large cores of tumors should be removed at time of biopsy to allow maximal amounts of tissue for the pathologist to work on. Additionally, with the routine use of special immunohistochemical staining techniques and sarcoma genetics testing, getting enough tissue is key.
In most cases, a core needle biopsy would be able to distinguish between a sarcoma and lymphoma. There are those very rare instances where tumors are very poorly differentiated, and even expert pathologists are not able to define the lineage of the cancer cells. I would say those instances are very rare.
B-cell lymphoma is a form of lymphoma.
Question 4: Elaborate more on Leiomyosarcoma
- Do you have treatment for leiomyosarcoma that metastasizes from uterine to lungs, skin and liver?
- What would be a recommended tracking monitor for a leiomyosarcoma survivor? The surgery conducted 2 years ago and followed by PET scan that indicated no suspicious cancers/ tumours in the body. Any recommendation to prevent this sarcoma to appear in the future? Thanks in advance.
- Do you have target medicine for leiomyosarcoma?
Leiomyosarcoma (LMS) is one of the most common forms of aggressive soft tissue sarcoma. It originates from the smooth muscle of the body. Unlike skeletal muscles, smooth muscles are muscles that we cannot control. Smooth muscles are present in the uterus (which contract and push the baby out of the mother’s womb), in blood vessels which control the amount of blood flow to an organ, in intestines to move intestinal contents down. In women, the 50% of LMS originate in the uterus. The other common site is in major blood vessels.
For LMS that has metastasized (spread) to the lungs, skin and liver, this is considered stage 4 disease. The aim of treatment in patients with stage 4 disease is to slow down the spread of disease; cure is unlikely with the current treatments we have right now. Treatment typically involves the use of chemotherapy. There are many agents approved for this indication. Sometimes radiation and or surgery can be considered in certain clinical circumstances.
For LMS survivors, typically patients who have completed treatment with no evidence of disease clinically and on scans, I would generally advise such patients to consider medical checks and periodic imaging studies to make sure there is no relapse of the LMS. As the years go by, the intervals between these medical checks could then be slowly lengthened.
Coming back to this question, if the LMS was operated 2 years ago without evidence of recurrence, then I would say this is a good sign. But continue your follow-up and imaging scans with your local doctors. Guidelines have suggested a surveillance strategy of 6-monthly consults with appropriate imaging studies until 5 years, at which time, consider yearly assessments.
Unfortunately there has been no consistent targetable mutation found in LMS. But we do have oral pazopanib approved for use in LMS and other aggressive soft tissue sarcoma. Simply put, Pazopanib is a medicine that attacks new blood vessel formation. Without new blood vessel formation, the tumors are not able to grow and multiple. Thus, Pazopanib is a form of targeted therapy, approved for use in LMS and other aggressive soft tissue sarcoma.
Question 5: Are trials underway for more advanced chemotherapy to target metastatic Osteosarcoma. immunotherapy trials and or targeted radiation
Osteosarcoma is a rare disease. As such, dedicated trials in this tumor type are not common. In the absence of dedicated clinical trials, one may also consider phase 1 trials (early stage trials) for participation should you be able to access them.
| POSTED IN | Cancer Treatments |
| TAGS | cancer survivorship, chemotherapy, immunotherapy, metastatic cancer, radiotherapy (radiation therapy), rare cancer, sarcoma, stage 4 cancer, targeted therapy, tumours |
| READ MORE ABOUT | Sarcoma |
| PUBLISHED | 02 September 2020 |