News & Articles

Sarcoma - A Complex, Aggressive Cancer: 4 Main Types
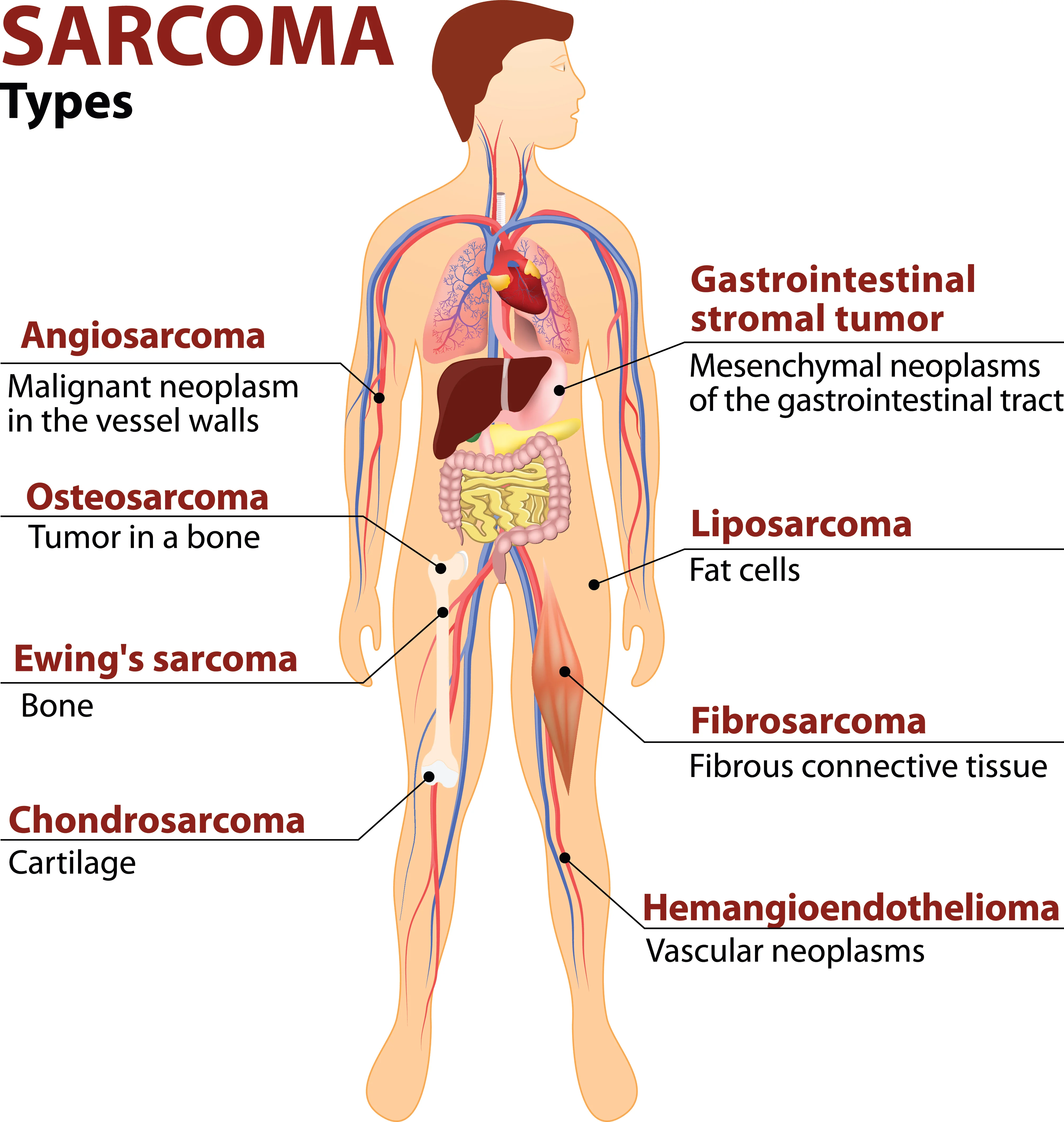
Dr Richard Quek from Parkway Cancer Centre shares about the main types of this tumour that grows in connective tissue.
Sarcomas are cancers which develop from the mesenchymal layer of our body.
In normal human development, amongst other structures, the mesenchymal layer develops to form connective tissue, fat cells, muscles, blood vessels and bones. As such, sarcomas may arise from various parts of the body, including skin, connective tissue, bones, blood vessels and even deep organs.
While less common than other cancers, sarcomas are generally aggressive.
Sarcomas are indeed a complex disease with more than 70 different subtypes reported. Broadly, they can be divided into bone and soft tissue tumours. More specifically, there are four main groups of sarcomas that require different methods of treatment:
1) Soft tissue sarcoma (STS)
Soft tissue sarcoma itself is a complex group of sarcomas, with more than 50 distinct subtypes. The more common subtypes include liposarcoma (fat cancers), leiomyosarcoma (muscle cancers), undifferentiated pleomorphic sarcoma (undifferentiated sarcoma) and angiosarcoma (blood vessel cancers).
A ubiquitous cancer, STS can develop from anywhere in the body: From the head and neck region, to the limbs, trunk and pelvis. It can affect organs deep inside the body, as well as superficially, on the skin. Sarcomas developing from the heart, blood vessels and scalp are also frequently seen.
As a disease, STS itself, is not common: It is seen in just five out of 100,000 people, though it affects younger people disproportionately. It is estimated that some 10 to 15 per cent of cases may be hereditary.
Sarcomas typically appear as a mass and may grow to be as large as 20cm or more. Depending on where it is located, it may cause different symptoms, such as a cough (if it is in the chest), limb or abdominal swelling. There may or may not be pain associated with the tumour masses.
STS is best treated with a combination of surgery, radiation and chemotherapy. After surgery is done to remove the sarcoma, radiation may be administered in cases where the tumour is high grade or large. Chemotherapy may be used to shrink the tumour before surgery, or to prolong life and alleviate the symptoms where the tumour has spread.
Traditional chemotherapeutic agents and oral targeted therapies are usually used to target the tumour, though ongoing trials have shown promise in immunotherapy drugs for some forms of STS.
The prognosis for those with STS is promising, with a good chance of cure or remission following treatment, even in cases where the cancer has spread. Regular reviews and scans are therefore critical.
2) Bone sarcomas
Bone sarcomas are cancers that originate primarily from the bones. The most common forms are osteosarcoma (bone), followed by chondrosarcoma (cartilage) and chordoma (notochord). Osteosarcomas tend to affect children and young adults, while chondrosarcomas and chordoma affect adults over a wide age range.
Symptoms include bone pain, bone swelling, and fractures after minimal trauma. Bone sarcomas can be detected through X-rays, MRIs, PET, CT and bone scans.
In the case of osteosarcoma, patients are usually treated with 10 to 12 weeks of chemotherapy, followed by surgery.
Wherever possible, amputation will be avoided to spare the affected limbs. This is then followed by further chemotherapy to eradicate all traces of the tumour. The use of chemotherapy is crucial and has been shown to triple the survival rate – from 20 per cent with surgery alone to 70 per cent with surgery and chemotherapy.
Following the end of treatment, close monitoring with disease surveillance is important. Should the disease recur, the lungs are the most likely organs to be affected. Hence imaging scans should always include the lungs.
Unlike other cancers where Stage 4 is rarely curable, surgery, even in advanced osteosarcoma which has spread, may be potentially curative in 30 per cent of patients. Such patients are well selected and usually have one or few lung lesions. Hence finding these lesions early when they are still small and amenable to surgery is important.
3) Gastrointestinal stromal tumours (GIST)
Gastrointestinal stromal tumour, also known as GIST, arises in the gastrointestinal tract. This form of sarcoma comes about as a result of a gene mutation, specifically in either the KIT or PDGFRA gene. The mutations cause uncontrolled cell growth, which in turn give rise to tumour formation.
Gastrointestinal stromal tumours are rare, found in as few as 15 per one million people.
This form of sarcoma can be treated very effectively with targeted therapies in the form of oral tyrosine kinase inhibitors (oral tablets), which help to prevent the cancer cell growth.
One of the first such therapies is imatinib, which has been consistently shown to shrink tumours in as many as two-thirds of all GIST patients. Before this drug came along, no effective therapy existed, and the survival rate for those with Stage 4 GIST was only about 18 months.
With the advent of imatinib, the median survival rate has risen tremendously to more than five years, even in patients with Stage 4 disease. Imatinib is well tolerated by many patients with few side effects. It is also convenient, being administered orally.
Imatinib has also been used to prevent GIST relapse following surgery (adjuvant treatment). Studies have shown that imatinib taken after surgery reduces the risk of relapse and improves survival in patients with high-risk disease.
Additionally, as imatinib is very effective in shrinking GISTs, it is sometimes administered prior to surgery to shrink large tumours so as to reduce the extent and morbidity of surgery. One example would be in large GISTs arising in the rectum where a morbid surgery with permanent stoma bag may be needed to clear the tumour.
In such cases, imatinib may be used to down-size the tumour, allowing for a more limited operation and thus saving the patient from a permanent stoma bag.
Due to rapid scientific advancements in the field of GIST, for those who have failed first-line imatinib treatment, there are now two additional treatments approved for use, in the form of sunitinib and regorafenib. Many more clinical trials are underway.
4) Ewing’s sarcoma (EWS) & Rhabdomyosarcoma (RMS)
Both Ewing’s sarcoma (EWS) and rhabdomyosarcoma (RMS) are highly aggressive sarcomas. But thankfully, they are also extremely sensitive to chemotherapeutic drugs. They can grow in the soft tissue and bone and arise from various parts of the body, such as the head and neck, spine or long bones, in the extremities and in the chest. These sarcomas tend to affect children and young adults.
Under the microscope, both EWS and RMS may look like, and sometimes be mistaken for, other cancers, so an accurate diagnosis is critical. Molecular testing may be needed to confirm diagnosis and help with prognosis as well.
Both EWS and RWS are particularly sensitive to chemotherapy and are typically treated with a combination of chemotherapy agents over an extended duration of nine to 12 months. Surgery and/or radiation are also included as part of the treatment protocol for the control of the local tumour.
Cure rates are high for EWS – the long-term survival rate in patients with localised EWS is 75 per cent, and even in patients where the cancer has spread, the long term survival rate is as high as 25 per cent. For RMS, survival rates depend on the subtype of the disease and age of patients.
| POSTED IN | Cancer Treatments |
| TAGS | primary bone cancer, sarcoma, stage 4 cancer, tumours |
| READ MORE ABOUT | Sarcoma |
| PUBLISHED | 02 August 2018 |