Sarcoma

Sarcomas are cancers which develop from the mesenchymal layer of our body. The mesenchymal layer develops to form connective tissue, fat cells, muscles, blood vessels and bones. As such, sarcomas may arise from various parts of the body, including skin, connective tissue, bones, blood vessels and even deep organs.
While less common than other cancers, sarcomas are generally aggressive. It is also a complex group of disease with more than 70 different subtypes reported. Broadly, they can be divided into bone and soft tissue tumours. More specifically, there are four main groups of sarcomas that require different methods of treatment:

Bone Sarcoma
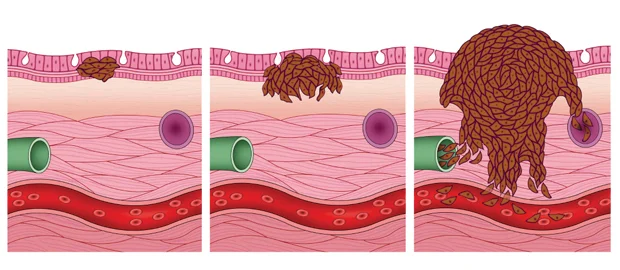
Bone sarcomas are cancers that originate primarily from the bones. The most common forms are osteosarcoma (bone), fol- lowed by chondrosarcoma (cartilage) and chordoma (notochord). Osteosarcomas tend to affect children and young adults, while chondrosarcomas and chordoma affect adults over a wide age range.
Soft Tissue Sarcoma (STS)
Soft tissue sarcomas comprise more than 50 distinct subtypes. The more common subtypes include liposarcoma (fat cancers), leiomyosarcoma (muscle cancers), undifferentiated pleomorphic sarcoma (undifferentiated sarcoma) and angiosarcoma (blood vessel cancers).
STS can develop from anywhere in the body: from the head and neck region, to the limbs, trunk and pelvis. It can affect organs deep inside the body, as well as superficially on the skin. Sarco- mas developing from the heart, blood vessels and scalp are also frequently seen.
Gastrointestinal Stromal Tumours (GIST)
Gastrointestinal stromal tumour, also known as GIST, arises in the gastrointestinal tract, most commonly in the stomach, but may also affect the small bowels and rectum. This form of sarcoma comes about as a result of a gene mutation, specifically in either the KIT or PDGFRA gene. These mutations cause uncontrolled cell growth, which in turn give rise to tumour formation.
Ewing’s Sarcoma (EWS) & Rhabdomyosarcoma (RMS)
Both Ewing’s sarcoma (EWS) and rhabdomyosarcoma (RMS) are highly aggressive sarcomas. They can grow from soft tissue or bones and arise from various parts of the body, such as the head and neck, spine or long bones, in the extremities and in the chest. These sarcomas tend to affect children and young adults.
Depending on the type of sarcoma, the factors causing it are different. For example, in GIST, genetic mutations in KIT or PDGFRA genes result in disease. In other sarcomas like Ewing’s sarcoma and rhabdomyosarcoma, genetic mutations (called translocations) are associated with these sarcomas. How and why these genetic mutations occur in tumours is however unknown. What we do know is that there are certain risk factors may raise the risk of developing sarcoma and they include:
- Family history of sarcoma
- Familial genetic disorders such as neurofibromatosis, Gardner syndrome, retinoblastoma, or Li-Fraumeni syndrome.
- Exposure to radiation
- Damaged lymphatic system
Patients with soft tissue sarcomas often have no symptoms until late in the course of illness. Depending on the type of sarcoma, signs and symptoms may also be different:
A lump - it may start small. It is usually painless and may grow to a large size over time (size of a small water melon).
Pain – more commonly affects patients with bone sarcomas. As patients are young and active, these bone pains are usually attributed to sports injury or growing pains resulting in delayed diagnosis. Pain at rest and at nights is an important warning symptom.
Intestinal symptoms – more often seen in GIST patients and patients whose sarcomas arise in the abdomen (such as liposarcoma). Symptoms include abdominal distention, bloatedness, pain, vomiting blood or blood in stools.
Organ specific symptoms – as sarcoma can arise from anywhere in the body, they may grow and compress on surrounding organs resulting in a wide range of symptoms including cough and breathlessness (chest sarcoma), vision problems (head and neck sarcoma).
If your doctor thinks you may have a sarcoma, you will probably need a full examination and tests, including:
- A sample of cells from the tumour, called a biopsy.
- Imaging tests, such as a CT scan, MRI or PET scan to help see the tumour and assess for spread of disease.
- A bone scan, may also be needed for some patients with osteosarcoma.
Multimodality treatment, using a combination of surgery, chemotherapy and/or radiation gives the best outcome in sarcoma. Specific treatment choice depends on
a) Type of sarcoma
b) Stage (extend of spread) of sarcoma
c) Location of sarcoma
In Bone Sarcoma
- In osteosarcoma, treatment typically consists of a program of chemotherapy (10-12 weeks) followed by surgery and then further chemotherapy post-surgery.
- In chondrosarcoma and chordoma, surgery alone remains the mainstay of treatment.
In Soft Tissue Sarcoma
- In localised disease, surgery is typically performed to remove the tumour with a cuff of normal tissue. Radiation before or after surgery is oftentimes considered for patients with large, deep and/or high grade sarcomas.
In GIST
- In localised disease, surgery is usually performed. A medicine called imatinib, that targets the KIT and PDGFRA mutation is highly effective and is given to patients with high risk disease.
- In advanced disease where surgery is not possible, sometimes imatinib can be given to down size the tumour, making surgery easier at a later date.
- For those whose GIST have spread, targeted therapy like imatinib, sunitinib and regorafenib etc have greatly improved the survival of patients to an average of more than 5 years now.
In Ewing’s sarcoma and rhabdomyosarcoma
- While in Ewing’s sarcoma and rhabdomyosarcoma, these tumours are exquisitely sensitive to chemotherapy, as such chemotherapy is typically given first prior to local treatment like surgery and/or radiation with further consolidation chemotherapy to follow later.
CanHOPE is a non-profit cancer counselling and support service provided by Parkway Cancer Centre, Singapore. CanHOPE consists of an experienced, knowledgeable and caring support team with access to comprehensive information on a wide range of topics in education and guidelines in cancer treatment.
CanHOPE provides:
- Up-to-date cancer information for patients including ways to prevent cancer, symptoms, risks, screening tests, diagnosis, current treatments and research available.
- Referrals to cancer-related services, such as screening and investigational facilities, treatment centres and appropriate specialist consultation.
- Cancer counselling and advice on strategies to manage side effects of treatments, coping with cancer, diet and nutrition.
- Emotional and psychosocial support to people with cancer and those who care for them.
- Support group activities, focusing on knowledge, skills and supportive activities to educate and create awareness for patients and caregivers.
- Resources for rehabilitative and supportive services.
- Palliative care services to improve quality of life of patients with advanced cancer.
The CanHOPE team will journey with patients to provide support and personalised care, as they strive to share a little hope with every person encountered.